
β Thalassemia
Hemolytic anemia due to deficient synthesis of β globin chains.
Molecular basis of β Thalassemia
majority of the mutations are point mutations. The mutations are of two types
βo mutations – absent β-globin synthesis
β+ mutations – reduced (but detectable) β-globin synthesis
POINT MUTATIONS
there are 3 major causes
1. Mutations affecting mRNA processing – Splicing mutations
a. Create an “ectopic” splice within an intron leading to Both normal and abnormal splicing. Hence Some normal beta globin mRNA is also made and thereby these are β+ mutations
b. Some destroy normal mRNA splicing junctions which Completely prevent the production of betaglobin mRNA synthesis ,leading to βo mutations
2. Transcription mutations – Promotor region mutations
these reduce transcription by 75 to 80%, Some normal beta globin Chains are made and hence these are β+ mutations
3. Mutations affecting mRNA translation – Chain terminator mutations. they can be either Nonsense mutation or Frameshift mutations, which finally Block translation, thereby Prevent beta globin synthesis and hence these are βo mutations
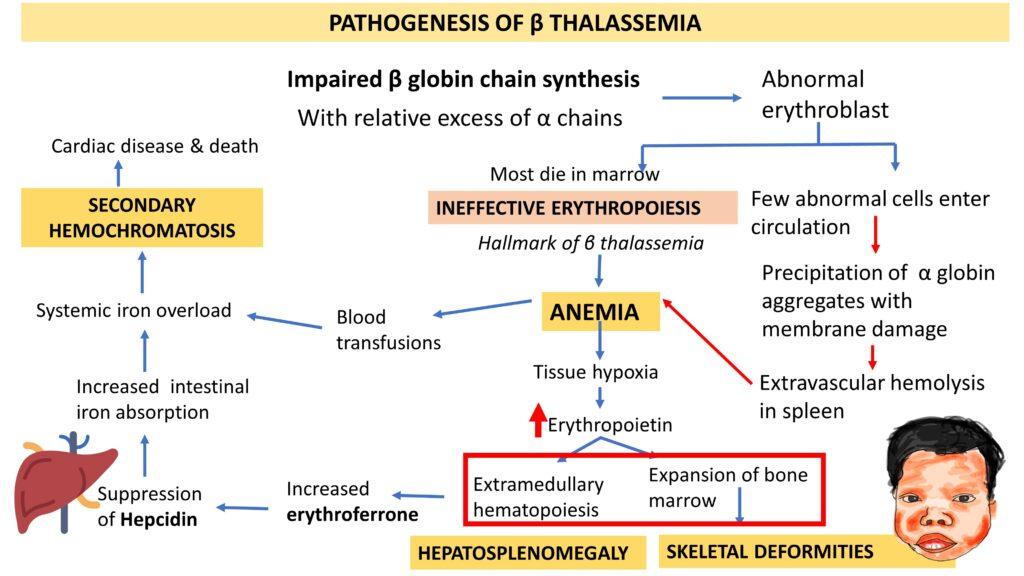
PATHOGENESIS OF β THALASSEMIA
The main mechanism is Impaired β globin chain synthesis and consequent relative excess of α chains leading to anemia, hepatosplenomegaly, skeletal deformities and secondary hemochromatosis as illustrated below.
Classification of clinical syndromes
β-thalassemia major. – individuals with two β-thalassemia alleles (β+/β+, β+/β0, or β0/β0) with severe, transfusion-dependent anemia
β-thalassemia minor /trait – Heterozygotes with one β-thalassemia gene and one normal gene (β+/β or β0/β) with mild asymptomatic microcytic anemia.
β-thalassemia intermedia – Independently of the genotype!Does not require transfusion or only sporadic transfusions
β-thalassemia major.
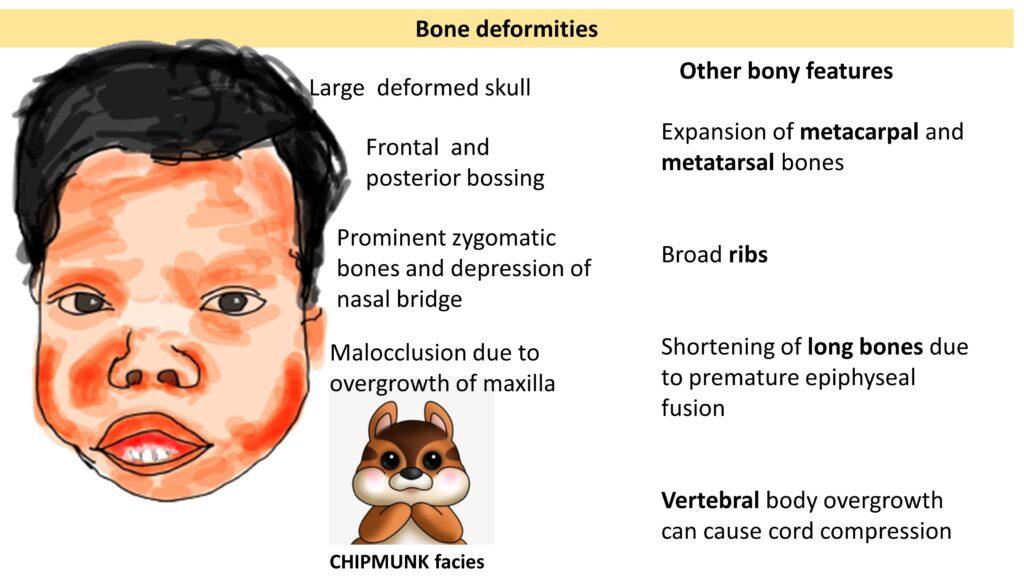
Also referred to as Mediterranean Anemia and Cooley’s anemia where Anemia usually appear in first year of life at the time when gamma chains are not replaced by beta chains. manifest with Pallor, splenomegaly, fever & failure to thrive along with skeletal deformities ( classical thalassemis /chipmunk facies)
chipmunk facies: Malocclusion due to overgrowth of maxilla with Prominent zygomatic bones and depression of nasal bridge, Frontal and posterior bossing and Large deformed skull
Hair–on end appearance/ Crew cut appearance – Suggests an extreme degree of medullary erythropoiesis and show long, thin vertical striations that resemble hairs standing on ends on x rays.

Lab Diagnosis of β Thalassemia Major
CBC- Markedly decreased hemoglobin, Low MCV & MCH
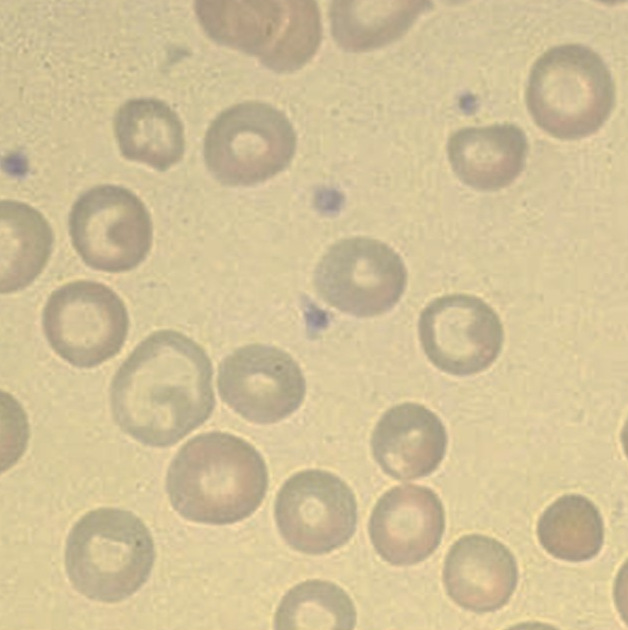
Peripheral smear- Marked anisopoikilocytosis, Microcytic hypochromic with many target cells.
Hb Electrophoresis – Elevated HbF. HbA2 levels are sometimes high but more often are normal or low.
Serum Iron – Elevated
Radiology – Bony features as described earlier!
Treatment
Blood transfusions
Iron chelation therapy if there is iron overload
Hematopoietic stem cell transplantation – curative!
β-thalassemia minor /trait
More common than beta thalassemia major. Usually asymptomatic or mild anemia
hypochromia, microcytosis, basophilic stippling, and target cells in peripheral smear along witn increase in HbA2 in Hemoglobin electrophoresis
Why it is important to recognize β-thalassemia minor /trait
1. It may be mistaken for iron deficiency: excluded by measurement of serum iron, total iron-binding
capacity, and serum ferritin
2. Implications for genetic counseling. Thalassemia is autosomal recessive disease
click here to watch the tutorial on beta thalassemia


{kind=link}