
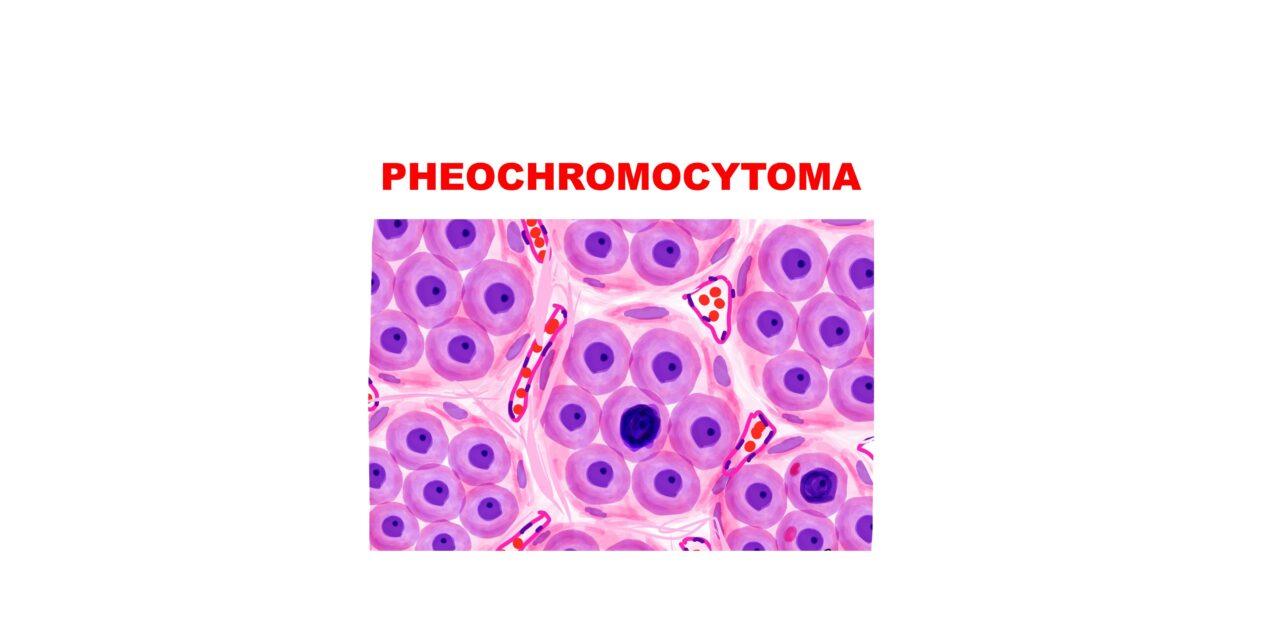
What is Pheochromocytoma, and where is it typically located?
Pheochromocytomas are neoplasms of the adrenal medulla composed of chromaffin cells, which can release catecholamines and sometimes peptide hormones, primarily affecting the body’s hormonal balance.
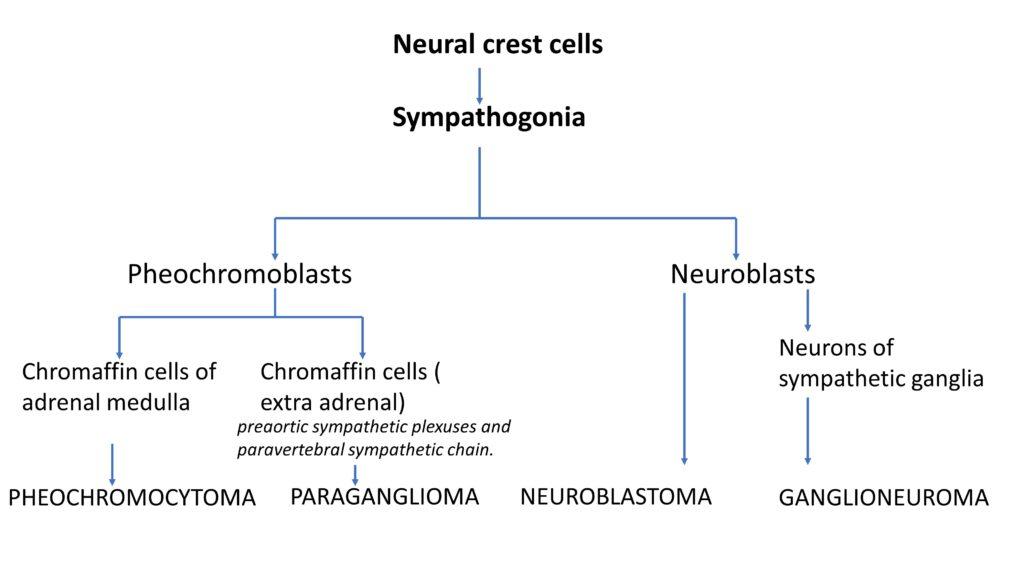
What is developmental lineage of neural crest cells
Neural crest cells differentiate into sympathogonia, which can further evolve into pheochromoblasts or neuroblasts. Pheochromoblasts give rise to chromaffin cells, which can form pheochromocytomas when located in the adrenal medulla or paragangliomas when these cells are extra-adrenal. Neuroblasts can mature into neurons of sympathetic ganglia or form neuroblastomas if they undergo neoplastic transformation. In some cases, neuroblastomas can mature into ganglioneuromas.
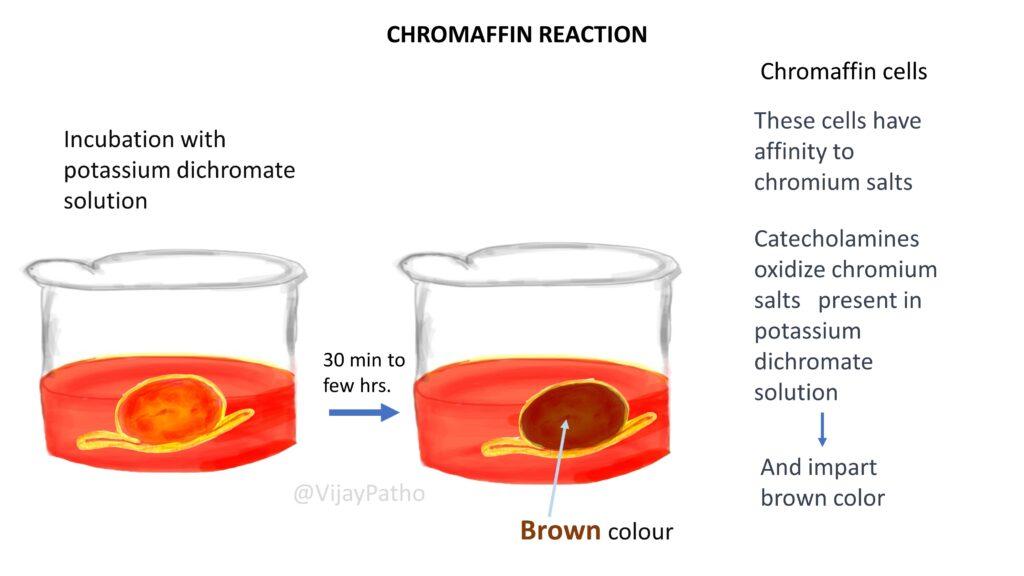
Describe the chromaffin reaction and its significance in diagnosing Pheochromocytoma.
The chromaffin reaction involves the affinity of chromaffin cells to chromium salts, leading to the oxidation of catecholamines, which imparts a brown color to the cells under a potassium dichromate stain, aiding in the diagnosis.
Explain the “Rule of 10s” in Pheochromocytoma.
Traditionally, the Rule of 10s suggests that approximately 10% of Pheochromocytomas are extra-adrenal, 10% are bilateral in sporadic cases, 10% are malignant, and 10% are not associated with hypertension. However, familial cases show variations in these percentages, presently it is around 25%.
What genetic mutations are associated with Pheochromocytoma, and how do they affect tumor growth?
Genetic mutations in genes such as RET, NF1, VHL, EPAS1, and the Succinate Dehydrogenase Complex (SDHB, SDHC, SDHD) can lead to uncontrollable cell growth and tumor development by affecting pathways like the Growth Factor Receptor Pathway and Hypoxia-Inducible Factors.
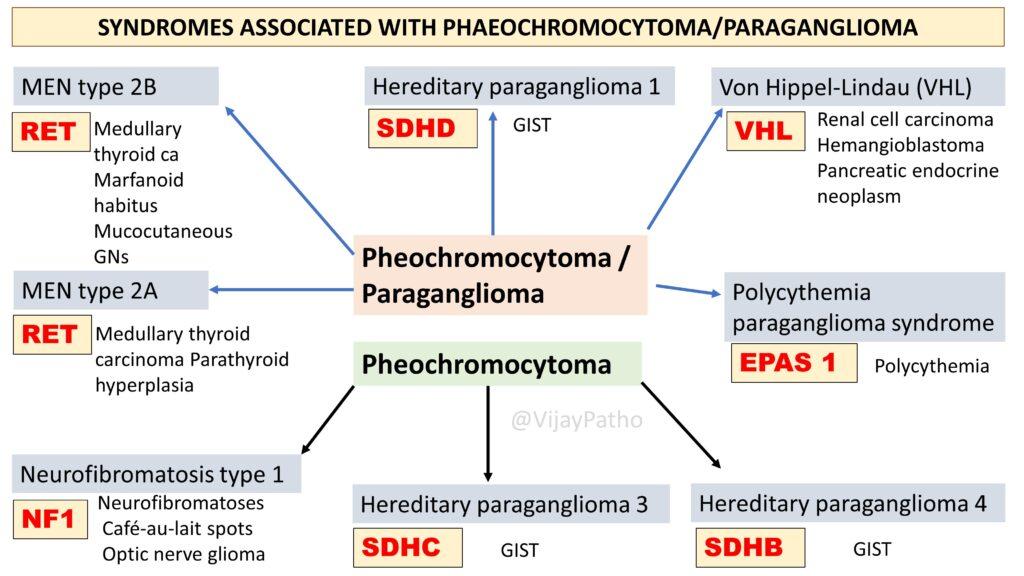
What are the genetic syndromes associated with pheochromocytoma?
Pheochromocytomas are often linked with several genetic conditions, such as Multiple Endocrine Neoplasia type 2A and 2B, Neurofibromatosis type 1, and hereditary paraganglioma syndromes. These conditions involve mutations in specific genes which can increase the risk of developing pheochromocytomas and also are associated with other lesions/tumors as illustrated below
How do Pheochromocytomas present morphologically?
Pheochromocytomas can range from small, circumscribed tumors to large masses with hemorrhagic, necrotic, and cystic changes. Microscopically, The characteristic is Zellballen pattern, or cell ball pattern, is observed, consisting of nests of chromaffin cells surrounded by sustentacular cells and a delicate vascular network.
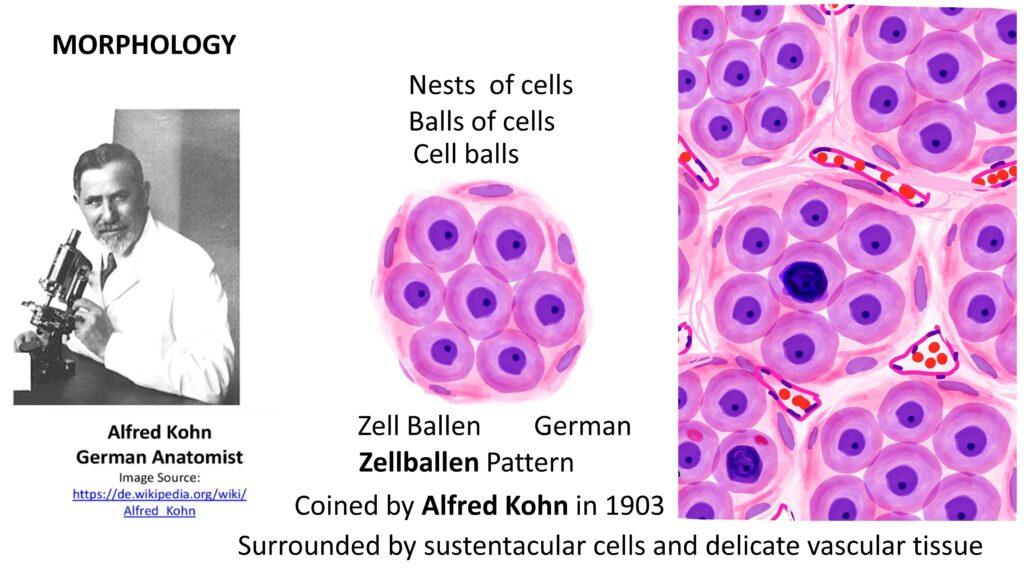
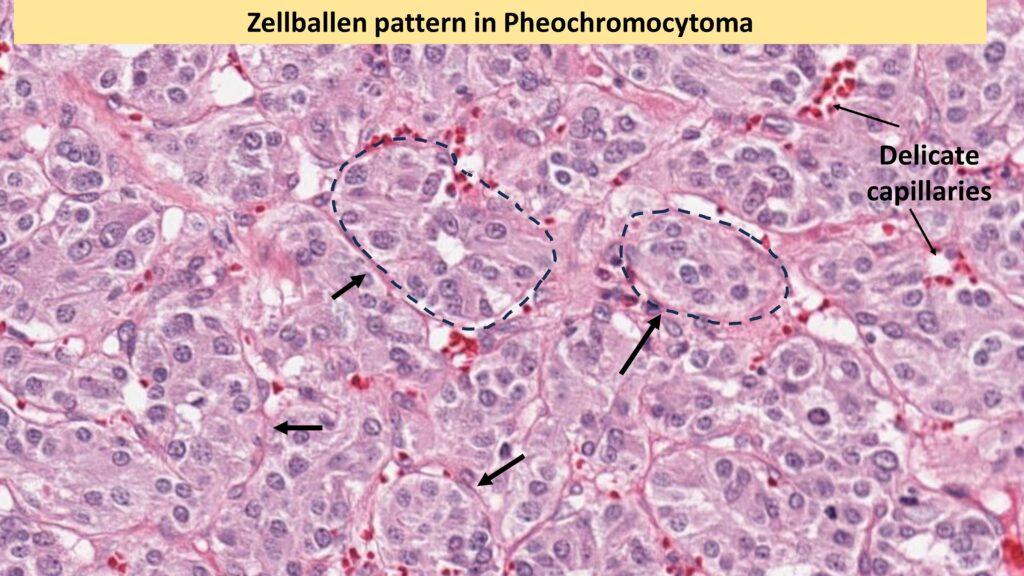
There can be significant amount of nuclear pleomorphism in pheochromocytoma. However, to render a diagnosis of Malignancy, once should demonstrate evidence of metastasis either ro regional lymph nodes or distant organs.
Discuss the clinical features and complications associated with Pheochromocytomas.
Hypertension is a dominant feature, with many patients experiencing paroxysmal episodes of severe blood pressure increases, accompanied by symptoms like tachycardia, palpitations, and headaches. Complications can include catecholamine-induced cardiomyopathy, congestive heart failure, pulmonary edema, myocardial infarction, ventricular fibrillation, and cerebrovascular accidents.
How does catecholamine cardiomyopathy develop in patients with pheochromocytoma?
Catecholamine cardiomyopathy is a result of the excessive secretion of catecholamines (epinephrine and norepinephrine) by pheochromocytomas, leading to direct toxicity on the myocardium or through constriction of myocardial blood vessels, ischemic damage, focal necrosis, mononuclear infiltrates, and interstitial fibrosis.
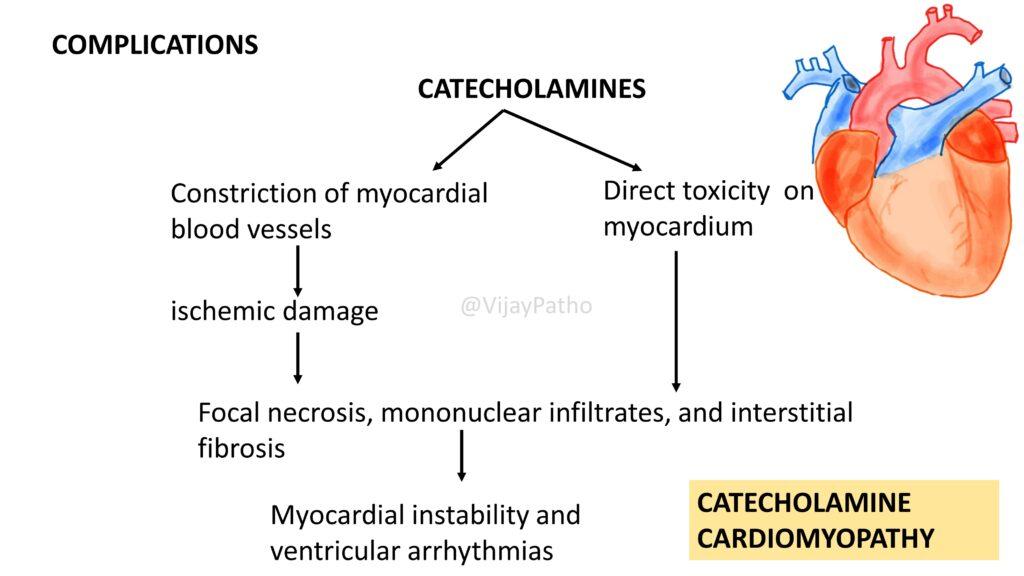
Clinically, catecholamine cardiomyopatyhy can precipitate severe cardiovascular complications such as myocardial instability, ventricular arrhythmias, congestive heart failure, pulmonary edema, myocardial infarction, and potentially life-threatening ventricular fibrillation.
Outline the diagnosis and treatment strategies for Pheochromocytoma.
Diagnosis involves demonstrating increased urinary excretion of free catecholamines, vanillylmandelic acid, and metanephrines.
Treatment typically involves surgical excision of isolated benign tumors, with preoperative and intraoperative management using adrenergic-blocking agents to prevent hypertensive crises. Multifocal lesions may require long-term medical management of hypertension.
CLICK below to watch the video tutorial on pheochromocytoma

{kind=link}