

What are neurodegenerative diseases?
Neurodegenerative diseases are characterized by the progressive loss of specific groups of neurons, which often have a shared function. These diseases involve the accumulation of protein aggregates, leading to a condition known as proteinopathy.
What causes protein aggregates to accumulate in neurodegenerative diseases?
Protein aggregates may accumulate due to mutations that alter protein conformation or disrupt protein processing and clearance pathways. Environmental or stochastic factors can also cause an imbalance between protein synthesis and clearance, leading to aggregation.
What are the types of protein aggregates found in neurodegenerative diseases?
Protein aggregates can be classified into two types based on size: large aggregates, which are non-toxic, and smaller, oligomeric aggregates, which are toxic to cells. Additionally, these aggregates are resistant to degradation and can induce further aggregation in neighboring cells, a characteristic feature of prions.
How are neurodegenerative diseases classified?
These diseases are classified based on the primary functional system affected. Categories include cortical degeneration (often manifesting as dementia), basal ganglia degeneration (leading to movement disorders), spinocerebellar degeneration (resulting in ataxia), and motor neuron degeneration (causing muscle weakness).

What are prion diseases, and how are they unique among neurodegenerative diseases?
Prion diseases are a type of neurodegenerative disorder characterized by the intercellular spread and aggregation of misfolded prion proteins, leading to rapidly progressive neurological decline. Unlike other proteinopathies, prion diseases are transmissible and can provoke additional protein aggregation in affected cells.
What is the pathogenesis of prion diseases?
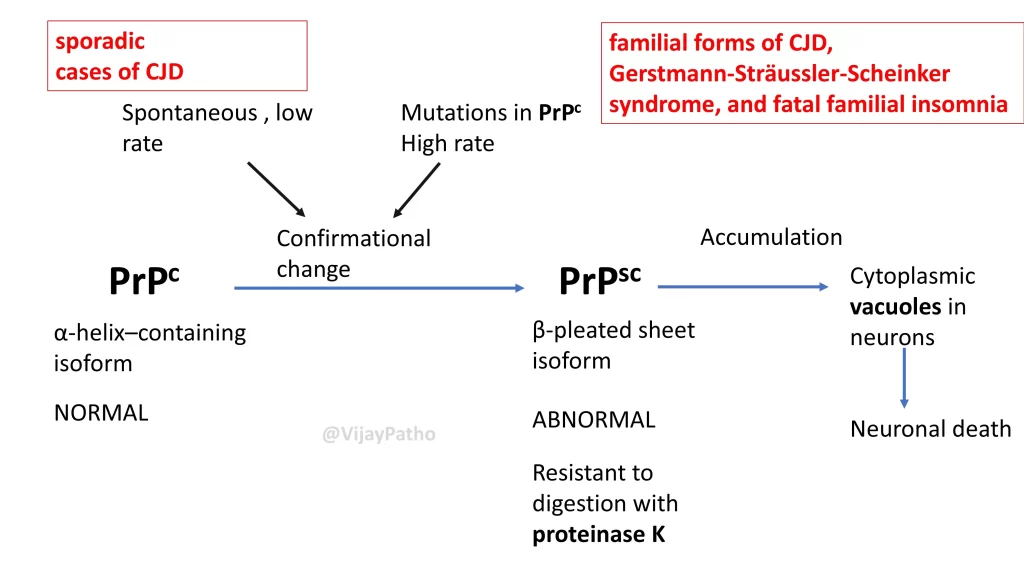
Prion diseases involve the transformation of normal cellular prion proteins (PrP^C) into a misfolded form known as PrP^Sc. This abnormal form is characterized by a higher content of beta-pleated sheets, making it resistant to enzymatic digestion and capable of promoting further misfolding and aggregation in host tissues.
What are some examples of prion diseases and their symptoms?
One of the most well-known prion diseases is Creutzfeldt-Jakob Disease (CJD), which is marked by rapidly progressive dementia and motor symptoms like myoclonus—a sudden, involuntary muscle jerk triggered by stimuli. Other prion diseases include Gerstmann-Sträussler-Scheinker syndrome and fatal familial insomnia.
How can prion diseases be transmitted?
Prion diseases can be transmitted iatrogenically through medical procedures involving the transplantation of infected tissues or the use of contaminated instruments. Some forms, like variant CJD, can also be acquired through the consumption of contaminated food.
What are the diagnostic hallmarks of Creutzfeldt-Jakob Disease (CJD)?
CJD is characterized by spongiform changes in the brain, which appear as vacuoles of varying sizes within the neuropil or around neurons. Advanced cases may show large cyst-like spaces, a condition referred to as status spongiosis. The presence of PrP^Sc can also be confirmed using immunohistochemical techniques.


Is there a treatment for prion diseases?
Currently, there is no effective treatment for prion diseases. The average survival time after symptom onset is about seven months, highlighting the rapid progression and severe impact of these disorders.
CLICK BELOW to watch the video tutorial on prion diseases


{kind=link}