
What are cardiomyopathies?
Cardiomyopathies are a group of diseases affecting the myocardium (heart muscle). They are associated with mechanical dysfunction (like poor pumping) and/or electrical dysfunction (like arrhythmias). These conditions often show inappropriate thickening (hypertrophy) or dilation of the heart chambers.
How are cardiomyopathies classified?
They are broadly classified into two types:
Primary cardiomyopathies: Involving only the heart.
Secondary cardiomyopathies: Involving the heart as part of a systemic or multi-organ disorder.
Primary cardiomyopathies can be further divided into genetic or acquired forms.
What causes cardiomyopathies at the molecular level?
Genetic mutations are a major cause. These mutations affect proteins involved in:
Energy production
Cardiac muscle contraction
Cell-to-cell connections
Cytoskeleton and extracellular matrix linkage
Mutations disrupt normal contraction, relaxation, and ion transport, leading to arrhythmias.
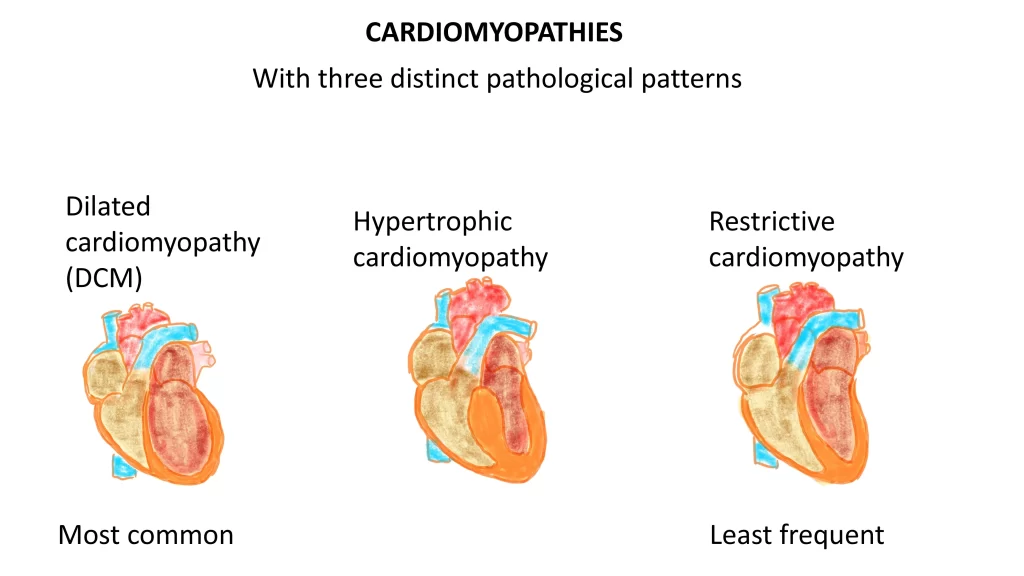
What are the three main types of cardiomyopathies?
Dilated cardiomyopathy (DCM) — most common
Hypertrophic cardiomyopathy (HCM)
Restrictive cardiomyopathy (RCM) — least common
What is dilated cardiomyopathy?
Dilated cardiomyopathy is characterized by:
Progressive enlargement (dilation) of all heart chambers
Weak heart contractions (systolic dysfunction)
Although dilation is the main feature, some degree of cardiac muscle thickening (hypertrophy) can also be present.
How is dilated cardiomyopathy diagnosed?
It is diagnosed by ruling out other causes like:
Ischemic heart disease
Valvular disease
Hypertension
Congenital heart disease
If no clear cause is found, it is labeled as primary or idiopathic dilated cardiomyopathy.
What are the genetic causes of dilated cardiomyopathy?
About 50% of dilated cardiomyopathy cases are familial. Mutations commonly affect:
Cytoskeletal proteins
Sarcolemmal proteins
Nuclear envelope proteins (like lamin A/C)
The most important mutation involves the titin protein (TTN truncation), seen in 10–20% of cases.
How is dilated cardiomyopathy inherited?
Most commonly autosomal dominant with variable penetrance
It can also be X-linked, autosomal recessive, or mitochondrial
Mutations in the dystrophin gene (responsible for muscular dystrophies) can also lead to dilated cardiomyopathy, often with conduction system abnormalities.
Besides genetic causes, what other factors cause dilated cardiomyopathy?
Other causes include:
Myocarditis (especially Coxsackie virus B infections)
Alcohol and toxins (ethanol and acetaldehyde)
Thiamine deficiency (beriberi heart disease)
Cobalt poisoning
Cardiotoxic chemotherapy
Peripartum cardiomyopathy (late pregnancy or postpartum, linked to anti-angiogenic factors like SFLT1)
Iron overload (seen in hemochromatosis or after repeated blood transfusions)
Supraphysiologic stress (persistent tachycardia, hyperthyroidism, catecholamine excess)
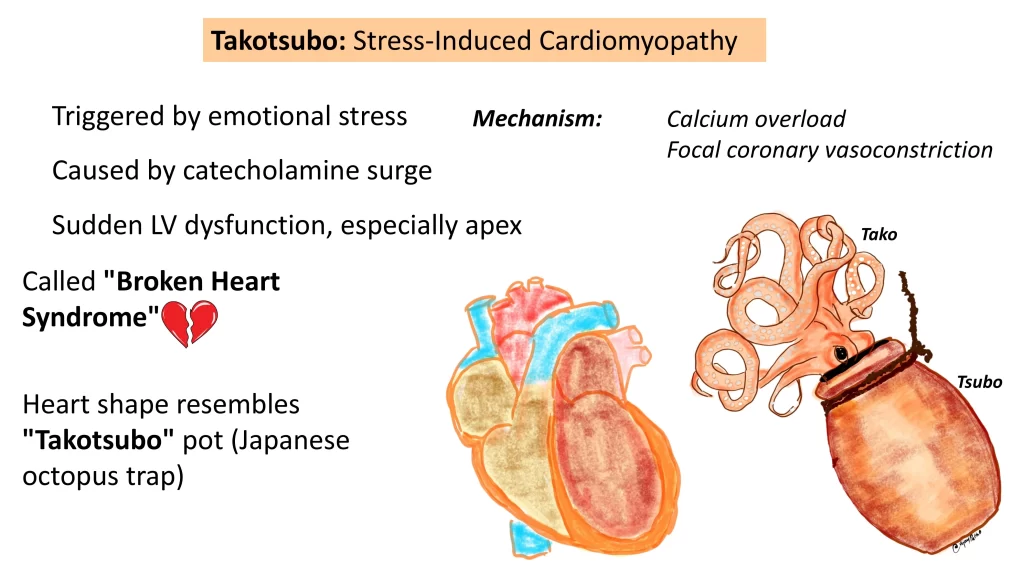
What is Takotsubo cardiomyopathy?
Also known as stress-induced cardiomyopathy or broken heart syndrome.
Triggered by emotional stress leading to a surge of catecholamines.
Results in sudden calcium overload at the apex of the heart, causing temporary left ventricular dysfunction.
The heart takes on the shape of a Japanese octopus trap (“Takotsubo”).
What are the morphological features of dilated cardiomyopathy?
Grossly:
The heart is enlarged, heavy, and flabby — about 2–3 times the normal weight.
All chambers are dilated.
In Takotsubo cardiomyopathy, the apex is predominantly involved.
Stasis of blood in the chambers can lead to mural thrombus formation and risk of thromboembolism.
Primary valvular changes are not seen. Any valvular dysfunction is usually secondary to chamber dilation.
Microscopy:
Non-specific findings
Variable interstitial and endocardial fibrosis
Subendocardial scars suggest prior ischemia
Enlarged and hypertrophied cardiac myocytes
Irregular, stretched, or distorted nuclei
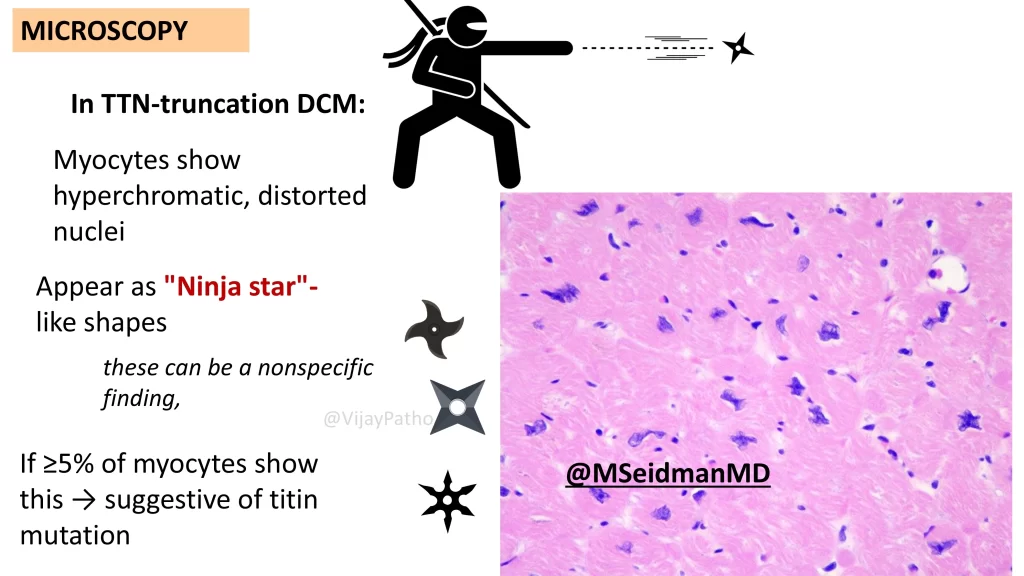
What is the special finding in TTN truncation dilated cardiomyopathy?
In cases with TTN truncation:
Myocytes show hyperchromatic, distorted nuclei with a “ninja star” appearance.
If these changes are seen in more than 5% of myocytes, it strongly suggests TTN mutation.
What are the clinical features of dilated cardiomyopathy?
Symptoms of congestive heart failure: shortness of breath, fatigue, reduced exercise tolerance
Secondary mitral regurgitation
Arrhythmias
Thromboembolism
The typical age of presentation is 20–50 years, but it can occur even in children.
What is the prognosis for patients with dilated cardiomyopathy?
High mortality: 10–15% per year
Death is often due to progressive heart failure or sudden arrhythmias
How is dilated cardiomyopathy treated?
Some patients respond to medications and biventricular pacing.
In advanced cases, cardiac transplantation is the definitive treatment.
Ventricular assist devices can improve long-term outcomes in selected patients.




{kind=link}