
Cardiomyopathies are a group of non-ischemic diseases of the myocardium. They are characterized by mechanical and/or electrical dysfunction of the heart. Morphologically, they may demonstrate abnormal ventricular hypertrophy or dilatation. They may have a genetic component. They may be either primary (heart-specific) or secondary (part of a broader systemic illness).
This discussion will be confined to the two most common entities namely, dilated cardiomyopathy (DCM) & hypertrophic cardiomyopathy (HCM)

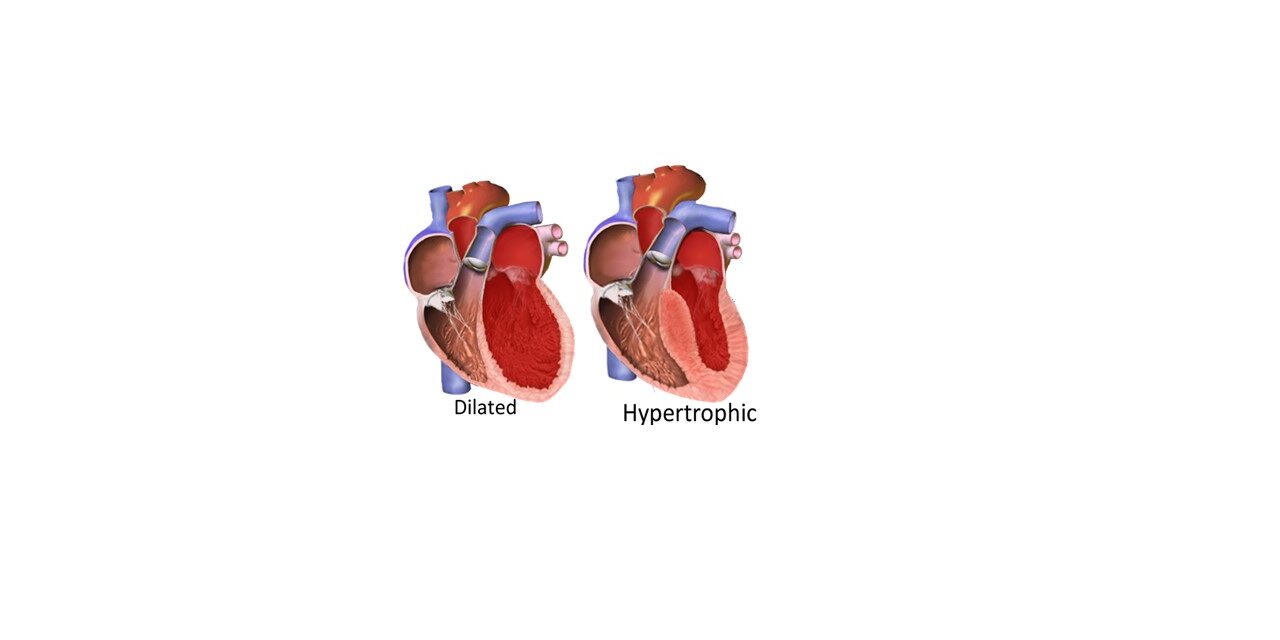
Dilated cardiomyopathy:
DCM can occur secondary to viral myocarditis or cardiotoxins in genetically predisposed individuals. Familial cases occur due to mutations in more than 50 genes encoding proteins in the sarcomeres, ion channels, cytoskeleton, nuclear envelope, and mitochondria of the cardiomyocytes.
Gross:
The heart is markedly enlarged and heavy with dilatation of all four chambers. The ventricular walls are of approximately normal thickness and can show varying degrees of fibrosis. The annulus of the mitral and tricuspid valves may be dilated.
Microscopy:
The histological features are non-specific. Variable degrees of cardiomyocyte hypertrophy and interstitial fibrosis may be present. Come of the cardiomyocytes may be attenuated, stretched or irregular.
Clinical features:
Inefficient contraction reduces the ejection fraction and eventually leads to congestive cardiac failure. Valvular regurgitation, abnormal cardiac rhythms due to fibrosis and intracardiac thrombi with emboli can occur, sometimes resulting in sudden death.
Hypertrophic cardiomyopathy:
HCM is a genetic disease due to mutations in numerous sarcomeric protein genes including beta-myosin heavy chain and myosin-binding protein C. The inheritance is autosomal dominant with variable expressivity, and incomplete, age-related penetrance. Modifier genes and environmental factors can affect the disease expression.
Gross:
HCM is characterized by increased thickness of the left ventricular wall without dilation of the chamber. The hypertrophy is characteristically asymmetric with disproportionate septal thickening. The papillary muscles may be hypertrophied or displaced. The mitral leaflet may be enlarged or elongated. Myocardial fibrosis may be seen.
Microscopy:
The cardiomyocytes are hypertrophied with abundant eosinophilic cytoplasm and box-shaped nuclei. They may show branching or whorling. The myocardial architecture is disorganised, with bundles of cardiomyocytes arranged at perpendicular and oblique angles to each other (myocardial disarray). The coronary arteries show medial hypertrophy. Interstitial or replacement fibrosis may be seen.
Clinical features:
HCM is characterised by multiple interrelated abnormalities, including left ventricular outflow tract (LVOT) obstruction, diastolic dysfunction, mitral regurgitation, myocardial ischemia, and arrhythmias.
LVOT obstruction may be due to systolic anterior motion (SAM) of the anterior mitral leaflet and mitral-septal contact, muscular obstruction due to hypertrophy or displacement of the papillary muscles. One third of patients have the non-obstructive form of HCM. Myocardial ischemia may occur due to medial hypertrophy and luminal narrowing of the coronary arteries.
While some individuals with HCM may be asymptomatic, others may present with cardiac symptoms. In a small number of affected individuals, the condition may be fatal.




{kind=link}