
HEMOGLOBINOPATHIES
Clinical diseases that result from a genetically determined abnormality of the STRUCTURE or SYNTHESIS of the hemoglobin molecule. The abnormality is associated with the globin chains. Note that the heme portion of the molecule is normal.
the globin abnormality is further classified into qualitative and quantitative defects.
QUALITATIVE :
Occurs as a result of genetic mutations involving globin protein chain
Aminoacid deletions or substitutions
These mutations cause structural variations of the globin chains
Hb S, Hb C, Hb M etc..
QUANTITATIVE
Occurs as a result of genetic defects that cause reduced synthesis of globin chains
The globin chains are structurally normal.
Collectively known as THALASSEMIAS
SICKLE CELL DISEASE
Sickle cell disease is a multisystem disorder that is caused by a single gene mutation.
Group of disorders characterized by production of sickle hemoglobin ( defective hemoglobin) HbS
they are classified into
1. SICKLE CELL ANEMIA – Homozygous
2. SICKLE CELL TRAIT – Heterozygous
3. OTHER SICKLING SYNDROMES – Compound Heterozygous
SICKLE CELL ANEMIA
Homozygous state caused by β-globin gene mutation.
Point mutation involving codon 6 of β-globin gene
This leads to the substitution of Valine residue for a glutamic acid residue. leading to sicking hemoglobin with abnormal solubility and stability resulting in SICKLING.
Why does sickling occur?
due to polymerization of HbS as illustrated below

What are the Factors affecting sickling ?
These include
Amount of other hemoglobin – HbA & HbF interferes with HbS polymerization – Decreases sickling.
Factors favoring sickling
HbC inhibits ion exchange across rbc membrane
Higher HbS results in increased MCHC
Transit time in microvasculature resultimg in stasis
Acidosis, fever, infections
Type of hemolysis in sickle cell anemia – EXTRAVASCULAR. Rarely INTRAVASCULAR when cells are fragile
CLINICAL FEATURES
Symptoms appear after 6 months as HbF is protective in first 6 months
Chronic hemolysis – anemia
Ischemic tissue damage resulting from occlusion of small blood vessels.
Chronic hyperbilirubinemia- Jaundice
What is “CRISIS” in sickle cell anemia?
it is a new episode developing quickly and includes
1. VASOOCCLUSIVE / PAIN CRISIS: Represents episodes of hypoxic injury and infarction associated with severe pain in the affected region. Most common involved sites are Bone , lungs , liver, brain , spleen and penis.
2. SEQESTRATION CRISIS: occur in children with intact spleen.
Massive sequestration of the sickle red cells leads to rapid splenic enlargement, hypovolemia & sometimes shock.
3. APLASTIC CRISIS : Transient cessation of BM erythropoiesis due to an acute infection of erythroid progenitor cells by Parvovirus B 19.Reticulocytes disappear from the peripheral blood, causing a sudden and rapid worsening of anemia.
4. HEMOLYTIC CRISIS – Marked increase in hemolysis leading to Sudden decrease in Hb
INFECTIONS IN SICKLE CELL ANEMIA
Susceptible to acute infections with encapsulated organisms due to Splenic hypofunction AND Defects in alternate complement pathway.
Pneumococcus pneumoniae and Haemophilus influenzae Septicemia and meningitis are most common cause of death in children
CHRONIC ORGAN DAMAGE in sickle cell anemia
SPLEEN – Initially splenomegaly, Gradual loss of splenic function due to infarct- AUTOSPLENECTOMY
BONE- Osteomyelitis
KIDNEY – Renal infarcts/papillary necrosis
SKIN- Ulcers
HEART – Ischemia , CCF due to long standing anemia
LAB FINDINGS IN SICKLE CELL ANEMIA
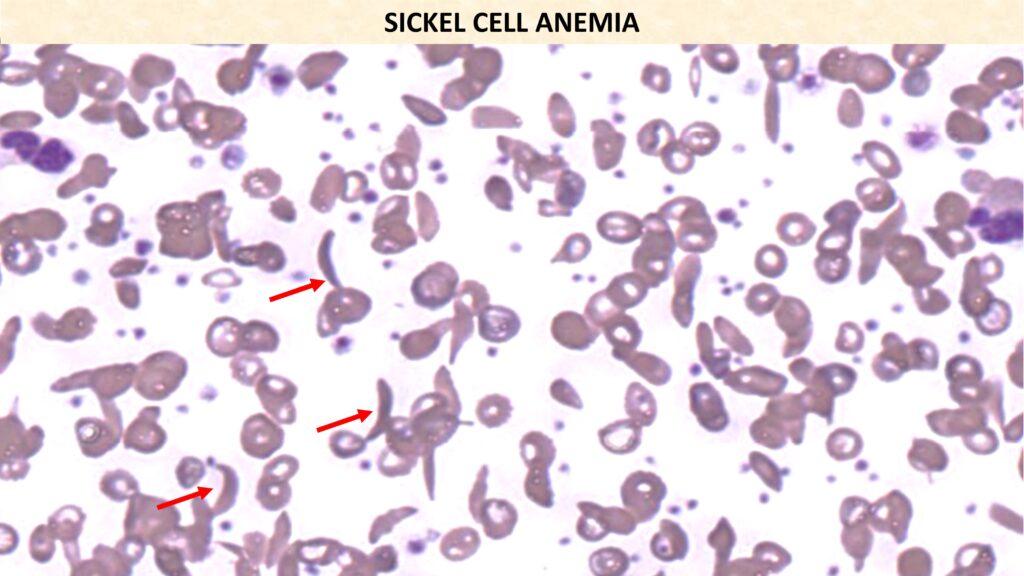
CBC &Peripheral Smear: Anisopoikilocytosis and presence of sickle cells.
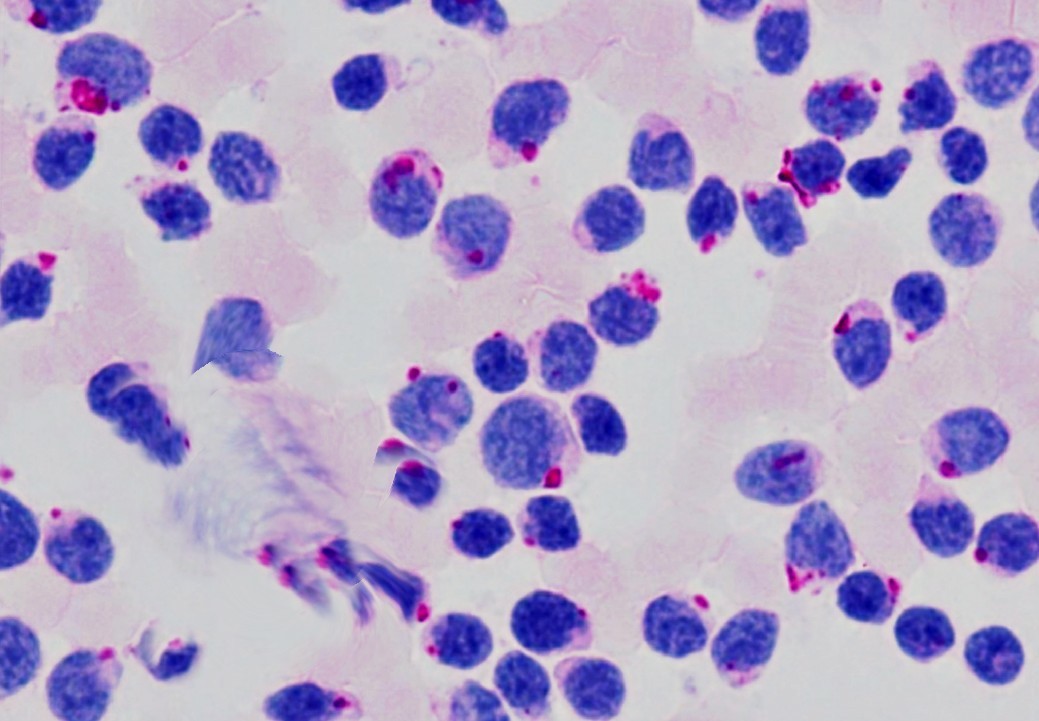
BONE MARROW – Compensatory erythroid hyperplasia
SERUM ; Reduced haptoglobin and elevated bilirubin, Iron ferritin & transferrin saturation
Hemoglobin electrophoresis to detect the presence of sickle cells.
DIAGNOSTIC/ CONFIRMATORY TESTS – High performance liquid chromatography
TREATMENT & Prognosis
90% of patients survive to 20 years of age
Around 50% survive beyond the fifth decade.
HYDROXYUREA, Inhibitor of DNA synthesis Increases red cell HbF levels and also has an anti-inflammatory effect
Decrease crisis related to vascular occlusions in both children and adults.
Hematopoietic stem cell transplantation- Chance for CURE!
GENE EDITING!!- Reverse hemoglobin switching, wherein stem cells produce RBCs that express HbF instead of HbS
CLICK HERE to watch the video tutorial on sickle cell anemia

{kind=link}